Die IR-Absorption von Kohlenmonoxid (CO) in Anhängigkeit vom Druck
Druckverbreiterung in der IR-Spektroskopie
Typischerweise ist die Größe der Absorption elektromagnetischer Strahlung vom Stoff, der Wellenlänge, Schichtdicke, Konzentration und bei Gasen auch der Temperatur und des Partialdruckes abhängig, somit von der Stoffmenge im Strahlengang.
Das Absorptionsverhalten von Gasen zeigt darüber hinaus auch eine weitere - deutliche Abhängigkeit - vom Gesamtdruck bzw. absolutem Druck.
Wird unter isothermen, isochoren und isobaren Bedingungen eine Küvette mit einem Gasgemisch, z.B CO in N2, gefüllt ist in reproduzierbarer Weise mit einem IR-Spektrometer eine bestimmte Extinktion zu messen. Wird nun der Innendruck der Küvette durch weiteres zuführen von CO-freiem Stickstoff (N2) langsam erhöht, so sinkt die relative Kohlenmonoxid-Konzentration zwar, die abs. Stoffmenge CO bzw. dessen Partialdruck bleibt jedoch konstant.
Da Stickstoff nicht IR-aktiv ist und die Stoffmenge CO konstant wird zunächst erwartet das die Absorption gleich bleibt. Bei Analysen ist mir jedoch aufgefallen das sich, trotz konstanter Stoffmenge CO, die Absorption ändert.
Die nachfolgende Grafik veranschaulicht den Response (Empfindlichkeitsverlauf, Flächenauswertung) der CO-Absorbtion durch Druckerhöhung mit reinem N2 in der Nähe des Backgrounddruckes.
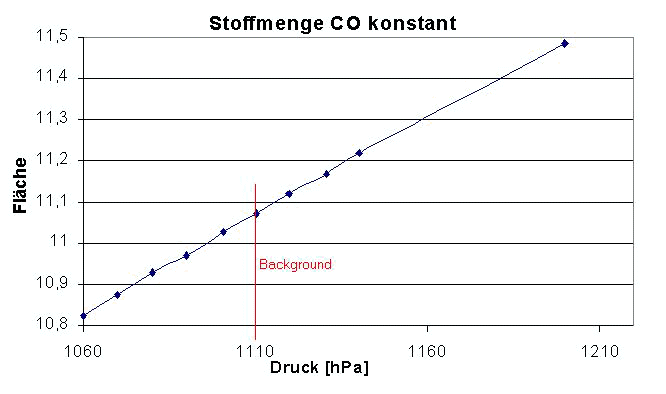
Um den Background herum scheint zunächst eine lineare Korrelation zu bestehen (s.o.)
Wird der zu betrachtende Druckbereich jedoch deutlich vergrößert ( s.u.) ist klar erkennbar das die Absorption von CO mit steigendem äußerem Zwang (Druckerhöhung) durch indifferente Matrix weiter zunimmt, Beobachtungen um den Backgrounddruck herum nicht als Messfehler zu werten sind und die Abhängigkeit eine Kurvenform ausweist.
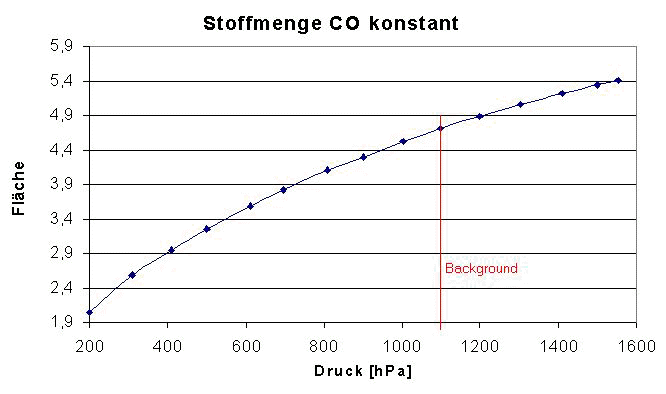
Auch bei starker Druckerhöhung, auf z.B. 5,5-fachem des Atmosphärendruck, ist die Druckverbreiterung klar zu sehen (s.u.)
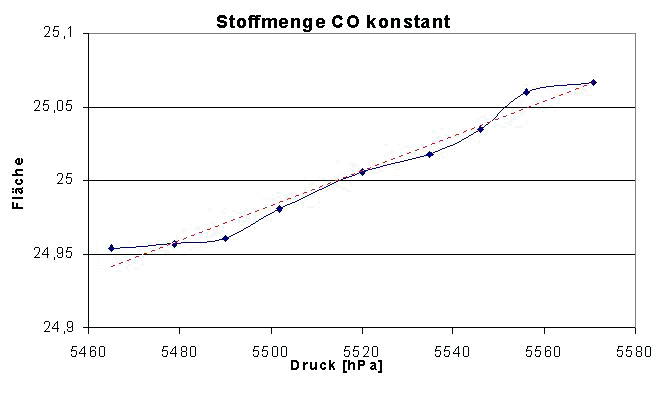
Ursache für dieses als Druckverbreiterung bezeichnetes Verhalten sind intermolekulare Wechselwirkungen (Impuls).
Bei entsprechender Auflösungen (ca. <2 Wellenzahlen) stellen sich Gasspektren nicht als eine breite Bande, sondern aufgelöst in viele Banden, da. Das Rotationsspektrum wird sichtbar. Ist die optische Auflösung des Spektrometers schlechter als die reale Bandenhalbwertsbreite kommt es zu einem Extinktionsverlust da die Datenpunkte nicht unbedingt auf dem realem Maximum liegen (Faltungseffekt). Kann die optische Auflösung des Spektrometers deutlich erhöht werden (<< 1 Wellenzahl) splittet sich die ungenügend aufgelöste Bande, bei div. Gasen, in z.T. mehrere feine Rotationsbanden auf bzw. erfaßte Einzelbanden werden nun Aufgrund dichterer Datenpunkte mit erhöhter Extinktion gemessen. Ist die opt. Auflösung des Spektrometers deutlich kleiner als die Bandenhalbwertsbreite, wird die betreffende Bande mit ihrem annähernd realen Maximum gemessen. Zu beachten ist dabei auch daß das Ausmaß der Druckverbreiterung neben dem Gesamtdruck auch von der Matrix abhängig ist. Selbst gleiche Gesamtdrücke bei unterschiedlichen Matrizes (z.B. N2, He, Ar, Luft ect.) können z.T. erhebliche Unterschiede verursachen!
Bei dispersiven Spektrometern liegt (lag?) die größte Unzulänglichkeit in einem nicht genügend auflösenden Spaltmaß. Bei FT-Spektrometern liegt sie neben der opt. Auflösung zudem noch in der math. Gewichtung (Apodisation) des Interferograms (Stichworte: Happ-Genzel, Boxcar-, Dreiecksapodisation ect.) und möglicher Interpolationsraten (Zerofilling). Da die Extinktionsverhältnisse sich nicht unbedingt proportional ändern kann die Gewichtung und Interpolation zudem noch einen Einfluß auf die Linearität haben, so daß es unabdingbar ist das Kalibrierkurven und Proben immer bei möglichst gleichen Bedingungen (Druck, Temperatur, Apodisation) aufgenommen werden. Selbst Abweichungen von nur wenigen hPa und Kelvin ließen sich meiner Erfahrung nach nicht einfach über die Gasgesetze umrechnen und ergaben z.T. noch Fehler in der Größenordnung von 1-2% rel. obwohl die Abweichungen für Druck (P) und Temperatur (T) sich in gleicher Größenordnungen befanden.
Bei dispersiven Spektrometern liegt die größte Unzulänglichkeit in einem nicht genügend auflösenden Spaltmaß. Bei FT-Spektrometern liegt sie neben der opt. Auflösung zudem noch in der math. Gewichtung (Apodisation) des Interferograms (Stichworte: Happ-Genzel, Boxcar-, Dreiecksapodisation ect.) und möglicher Interpolationsraten (Zerofilling). Da die Extinktionsverhältnisse sich nicht unbedingt proportional ändern kann die Gewichtung und Interpolation zudem noch einen Einfluß auf die Linearität haben, so daß es unabdingbar ist das Kalibrierkurven und Proben immer bei möglichst gleichen Bedingungen (Druck, Temperatur, Apodisation) aufgenommen werden. Selbst Abweichungen von nur wenigen hPa und Kelvin ließen sich meiner Erfahrung nach nicht einfach über die Gasgesetze umrechnen und ergaben z.T. noch Fehler in der Größenordnung von 1-2% rel. obwohl die Abweichungen für P und T sich in gleicher Größenordnungen befanden.
Überdies sei darauf hingewiesen das, nicht nur in der IR-Spektroskopie, die Linearität bzgl. des Lambert-Beer'schen Gesetzes stark komponentenabhängig sein kann.
Weitere Messdiagramme zur Druckverbreiterung von Kohlenmonoxid (CO) mit den Matrizes Helium (He), Argon (Ar) und Stickstoff; sowie Stickstoffmonoxid (NO) bei versch. Konzentrationen in Stickstoffmatrix, können Sie auf der folgenden Seite sehen.
